

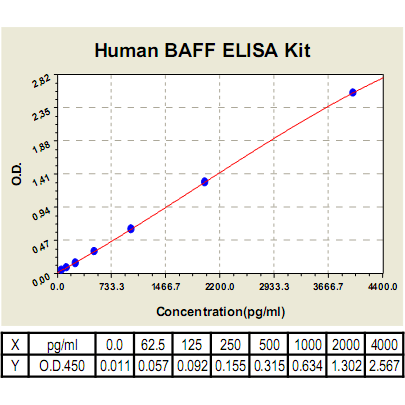
BAFF ELISA Kit, Human |
 |
BACKGROUND B cell activating factor (BAFF) and its close relative a proliferation-inducing ligand (APRIL) are members of the tumor necrosis factor (TNF) superfamily. Through interactions with their receptors, these two cytokines mediate the behavior of most B cells. Among the three receptors, BAFF receptor (BAFF-R) interacts solely with BAFF and is the dominant receptor governing primary B cell survival. In contrast, transmembrane activator and calcium modulator and cyclophylin ligand interactor (TACI) and B cell maturation antigen (BCMA) can interact with both APRIL and BAFF. BAFF interactions with BAFF-R control primary B cell selection, differentiation, and survival, as evidenced by the B cell hyperplasia and autoimmunity when BAFF is overexpressed or exogenously administered. BAFF depletion leads to a considerable decrease in mature B cells, without apparent effect on B cell genesis. In addition, the appropriate initiation and resolution of germinal center reactions, isotype switching, and plasma cell survival are influenced by BAFF or APRIL, presumably reflecting altered patterns of BAFF family receptors and mediators within antigen experienced B cell subsets.1
As with most TNF receptors lacking death domains, all three BAFF-binding receptors can engage at least one of the six known TNF-receptor-associated factors (TRAFs). Thus, BAFF-R contains a binding site for TRAF3 only, whereas TACI has sites for TRAF2, TRAF5, and TRAF6 and BCMA has sites for TRAF1, TRAF2, and TRAF3. Studies had already implicated TRAF2 and TRAF3 in B cell survival, through mechanisms influencing activation of both classical and nonclassical NF-κB transcription-factor pathways, as well as protein kinase C-δ (PKC-δ) retention. TRAF2 and TRAF3 interact to block p100 degradation, presumably through interactions with NIK. The binding of BAFF to BAFF-R interrupts this negative regulatory loop by sequestering and degrading TRAF3, thus allowing NIK accumulation, p100 degradation, and the subsequent nuclear localization of transcriptionally active heterodimers of p52 and RelB.2 Moreover, interaction of BAFF-R with BAFF leads to activation of the PI3K/Akt, ERK, and Pim-2 signaling pathways via IKKα, apparently via mechanisms independent of transcriptional activity of the NF-κB pathway. Phosphoinositide-dependent kinase-1 (PDK1) and PKCβ are also implicated in Akt activation in response to BAFF. Following activation, Akt targets the phosphorylation and cytoplasmic sequestration of the Forkhead transcription factor FOXO3a, which, when in the nucleus, targets the expression of the proapoptotic gene Bim. ERK activity further reduces cellular levels of the Bim protein by phosphorylation and degradation. Concomitantly, Akt induces transcriptional induction of the antiapoptotic gene Mcl-1 and blocks the inhibition of protein translation by 4E-BP1, including that of the Mcl-1 protein. Akt/mammalian target of rapamycin (mTOR) and Pim-2 signaling implements this inhibition of 4E-BP1 in response to BAFF. In addition to skewing the balance toward prevalence of antiapoptotic proteins, Akt plays a critical role in cellular growth and anabolism via the rapamycin-sensitive mTOR pathway. Another mechanism through which BAFF interaction with BAFF-R prevents cell death is by blocking the entry of proapoptotic PKCδ to the nucleus. Thus, BAFF regulates B cell survival and growth by preventing apoptosis at multiple levels and by mobilizing a major cellular metabolic pathway involving mTOR under Akt control.3
As with most TNF receptors lacking death domains, all three BAFF-binding receptors can engage at least one of the six known TNF-receptor-associated factors (TRAFs). Thus, BAFF-R contains a binding site for TRAF3 only, whereas TACI has sites for TRAF2, TRAF5, and TRAF6 and BCMA has sites for TRAF1, TRAF2, and TRAF3. Studies had already implicated TRAF2 and TRAF3 in B cell survival, through mechanisms influencing activation of both classical and nonclassical NF-κB transcription-factor pathways, as well as protein kinase C-δ (PKC-δ) retention. TRAF2 and TRAF3 interact to block p100 degradation, presumably through interactions with NIK. The binding of BAFF to BAFF-R interrupts this negative regulatory loop by sequestering and degrading TRAF3, thus allowing NIK accumulation, p100 degradation, and the subsequent nuclear localization of transcriptionally active heterodimers of p52 and RelB.2 Moreover, interaction of BAFF-R with BAFF leads to activation of the PI3K/Akt, ERK, and Pim-2 signaling pathways via IKKα, apparently via mechanisms independent of transcriptional activity of the NF-κB pathway. Phosphoinositide-dependent kinase-1 (PDK1) and PKCβ are also implicated in Akt activation in response to BAFF. Following activation, Akt targets the phosphorylation and cytoplasmic sequestration of the Forkhead transcription factor FOXO3a, which, when in the nucleus, targets the expression of the proapoptotic gene Bim. ERK activity further reduces cellular levels of the Bim protein by phosphorylation and degradation. Concomitantly, Akt induces transcriptional induction of the antiapoptotic gene Mcl-1 and blocks the inhibition of protein translation by 4E-BP1, including that of the Mcl-1 protein. Akt/mammalian target of rapamycin (mTOR) and Pim-2 signaling implements this inhibition of 4E-BP1 in response to BAFF. In addition to skewing the balance toward prevalence of antiapoptotic proteins, Akt plays a critical role in cellular growth and anabolism via the rapamycin-sensitive mTOR pathway. Another mechanism through which BAFF interaction with BAFF-R prevents cell death is by blocking the entry of proapoptotic PKCδ to the nucleus. Thus, BAFF regulates B cell survival and growth by preventing apoptosis at multiple levels and by mobilizing a major cellular metabolic pathway involving mTOR under Akt control.3
REFERENCES
1. Ota, M. et al: J. Immunol. 185:4128-36, 2010
2. Moisini, I. & Davidson, A.: Clin. Exp. Immunol. 158:155-63, 2009
3. Davidson, A.: Curr. Opin. Immunol. 22:732-9, 2010
2. Moisini, I. & Davidson, A.: Clin. Exp. Immunol. 158:155-63, 2009
3. Davidson, A.: Curr. Opin. Immunol. 22:732-9, 2010
Products are for research use only. They are not intended for human, animal, or diagnostic applications.
Параметры
Cat.No.: | CL0663 |
Target Protein Species: | Human |
Range: | 62.5pg/ml-4000pg/ml |
Specificity: | No detectable cross-reactivity with any other cytokine. |
Storage: | Store at 4°C. Use within 6 months. |
ELISA Kits are based on standard sandwich enzyme-linked immunosorbent assay technology. Freshly prepared standards, samples, and solutions are recommended for best results.
Документы

Информация представлена исключительно в ознакомительных целях и ни при каких условиях не является публичной офертой



