

Anti-PNPLA6: Rabbit Patatin-like Phospholipase Domain Containing Protein 6 Antibody |
 |
BACKGROUND The human genome expresses nine Patatin-like phospholipase domain containing proteins (PNPLA1–9). Members of this family share a protein domain discovered initially in Patatin, the most abundant protein of the potato tuber. Patatin is a lipid hydrolase with an unusual folding topology that differs from classical lipases.1 Mammalian PNPLAs include lipid hydrolases with specificities for diverse substrates such as triacylglycerols, phospholipids, and retinol esters. PNPLAs were found to serve critical roles in diverse aspects of lipid metabolism/remodeling and signaling. Several gain- and loss-of-function mouse models revealed important implications for PNPLA proteins in physiological processes. Moreover, the phenotypic consequences of mutations and gene polymorphisms in humans identified enzyme family members as key players in disease states linked to energy homeostasis, neuronal integrity, and age-related bone morphology.2,3
PNPLA6 is commonly termed neuropathy target esterase (NTE). Murine PNPLA6/NTE is primarily expressed in the nervous system, and the protein localizes to the endoplasmic reticulum (ER) and Golgi apparatus. PNPLA6/NTE exhibits in vitro esterase and hydrolase activity against membrane lipids, preferentially lysophospholipids. Importantly, the capacity of PNPLA6/NTE to hydrolyze lysophosphatidylcholine (LPC) in mouse brain hints at its conceivable physiological substrate. PNPLA6/NTE was originally identified as a target enzyme for the poisonous effect of organophosphates. These compounds cause a severe neurological disorder in vertebrates known as organophosphate induced delayed neuropathy, characterized by degeneration of long axons in the spinal cord and peripheral nerves, leading to paralysis of the lower limbs. In mice, global PNPLA6/NTE deficiency is embryonically lethal. Brain-specific deletion of PNPLA6/NTE in mice resulted in severe neuropathologic symptoms concomitant with disruption of the ER, vacuolation of nerve cell bodies, and abnormal reticular aggregates. In humans, PNPLA6/NTE mutations near the catalytic site were found to cause the severe PNPLA6/NTE -related motor neuron disease, which is characterized by progressive spastic paraplegia and distal muscle wasting. Thus, the lysophospholipase activity of PNPLA6/NTE has a critical function in maintaining axonal integrity across species.4,5
PNPLA6 is commonly termed neuropathy target esterase (NTE). Murine PNPLA6/NTE is primarily expressed in the nervous system, and the protein localizes to the endoplasmic reticulum (ER) and Golgi apparatus. PNPLA6/NTE exhibits in vitro esterase and hydrolase activity against membrane lipids, preferentially lysophospholipids. Importantly, the capacity of PNPLA6/NTE to hydrolyze lysophosphatidylcholine (LPC) in mouse brain hints at its conceivable physiological substrate. PNPLA6/NTE was originally identified as a target enzyme for the poisonous effect of organophosphates. These compounds cause a severe neurological disorder in vertebrates known as organophosphate induced delayed neuropathy, characterized by degeneration of long axons in the spinal cord and peripheral nerves, leading to paralysis of the lower limbs. In mice, global PNPLA6/NTE deficiency is embryonically lethal. Brain-specific deletion of PNPLA6/NTE in mice resulted in severe neuropathologic symptoms concomitant with disruption of the ER, vacuolation of nerve cell bodies, and abnormal reticular aggregates. In humans, PNPLA6/NTE mutations near the catalytic site were found to cause the severe PNPLA6/NTE -related motor neuron disease, which is characterized by progressive spastic paraplegia and distal muscle wasting. Thus, the lysophospholipase activity of PNPLA6/NTE has a critical function in maintaining axonal integrity across species.4,5
REFERENCES
1. Wilson, P.A. et al: J. Lipid Res. 47:1940-7, 2006
2. Kienesberger, P.C. et al: J. Lipid Res. 50(Suppl):S63-8, 2009
3. Zechner, R. et al: Curr. Opin. Lipidol. 16:333-40, 2005
4. Winrow, C.J. et al: Nat Genet. 33:477-85, 2003
5. van Tienhoven, M. et al: J. Biol. Chem. 277:20942-8, 2002
2. Kienesberger, P.C. et al: J. Lipid Res. 50(Suppl):S63-8, 2009
3. Zechner, R. et al: Curr. Opin. Lipidol. 16:333-40, 2005
4. Winrow, C.J. et al: Nat Genet. 33:477-85, 2003
5. van Tienhoven, M. et al: J. Biol. Chem. 277:20942-8, 2002
Products are for research use only. They are not intended for human, animal, or diagnostic applications.
Параметры
Cat.No.: | CA1226 |
Antigen: | Short peptide from human PNPLA6 sequence. |
Isotype: | Rabbit IgG |
Species & predicted species cross- reactivity ( ): | Human, Mouse, Rat |
Applications & Suggested starting dilutions:* | WB 1:1000 IP n/d IHC n/d ICC n/d FACS n/d |
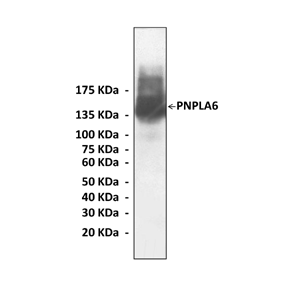
Predicted Molecular Weight of protein: | 145 kDa |
Specificity/Sensitivity: | Detects endogenous levels of PNPLA6 proteins without cross-reactivity with other related proteins. |
Storage: | Store at -20°C, 4°C for frequent use. Avoid repeated freeze-thaw cycles. |
*Optimal working dilutions must be determined by end user.
Документы
|
| ||||||||||
| ||||||||||

Информация представлена исключительно в ознакомительных целях и ни при каких условиях не является публичной офертой



