

Anti-p16INK4a: Rabbit p16INK4a Antibody |
 |
BACKGROUND Under normal circumstances, cell cycle initiation and progression is cooperatively regulated by several classes of cyclin-dependent kinases (CDKs), whose activities are in turn regulated by CDK inhibitors (CDKIs). To allow cell cycle progression, retinoblastoma protein (pRB) is phosphorylated by a holoenzyme complex containing cyclin D and a cyclin-dependent kinase (CDK4 or CDK6). The INK4 family of CDKIs comprises a number of small proteins — p16INK4a, p14ARF (murine p19ARF), p15INK4b, and p18INK4c — all of which have a role in cell cycle regulation. p14ARF (murine p19ARF) resides on chromosome 9p21 in humans and chromosome 4 (42.7) in mice, the INK4a/ARF locus encodes 2 different proteins, p16INK4a and p14ARF, via an alternative splicing mechanism. Lack of either protein predisposes the organism to the development of malignancy, though neither protein is required for normal growth and development. p14ARF and p16INK4a have long been recognized as mediators of senescence. p16INK4a binds and induces an allosteric conformational change in CDK4/CDK6 that inhibits the binding of ATP and substantially reduces the formation of the CDK4/6–cyclin D interface, which leads to the disruption of the interaction with D-type cyclins. This antagonizes cyclin binding and activation of CDK, thereby maintaining pRB in its hypophosphorylated and growth-suppressive state and induces G1 cell cycle arrest. Rb exists predominantly in an under-phosphorylated form that acts as a negative regulator of the cell cycle by sequestering transcription factors such as E2F. In the hyperphosphorylated state, Rb releases these factors, enabling them to activate transcription and drive the cell into S phase. In contrast to p16INK4a, p14ARF has a major role in the pathway involving p53, a transcription factor that regulates several genes involved in cell cycle checkpoints, stress responses, DNA damage and repair, and apoptosis. p14ARF binds the mouse double minute 2 (MDM2) protein and inhibits MDM2-mediated degradation of p53, which thus results in stabilization of p53. One of the important targets of p53 is the CDKI p21 Cip1/Waf1, which inhibits the activity of several cyclin-CDK complexes, thus arresting cells in both G1 and G2/M stages of the cell cycle.1 In addition, both p16INK4a and p14ARF play important role in aging. The accumulation of persistent and increasing DNA damage in senescent cells in response to telomere shortening, DNA damage, inappropriate activation of signaling pathways, and production of ROS during aging results in transcriptional activation of the INK4a/ARF locus. Mitogen stimulation may amplify the signals of DNA damage. Alternative stochastic mechanisms of aging that lead to p16INK4a/ p14ARF induction might also exist. Upregulation of p16INK4a and ARF activates pRB and p53 pathways, which in turn lead to cell cycle arrest and regenerative defects. In addition, p16INK4a/ p14ARF upregulation might influence cellular functions in mitotically inactive organs during aging.2
At the level of transcription, the expression of p16INK4a is modulated by 3 principal regulators: ETS1, inhibitor of DNA binding 1 (ID1), and B lymphoma Mo-MLV insertion region (BMI1). The p16INK4a appears to be tightly regulated in a feedback loop with Rb. When Rb is deleted, inactivated or hyperphosphorylated, p16INK4a transcription is stimulated, presumably through a feedback mechanism involving the release of Rb-sequestered transcription factors, driving the signal to shut down cell growth. The p16INK4a /CDK/cyclin/Rb/E2F regulatory cycle exhibits exquisite control on cell growth, and any alterations that upset this balance may lead to a deregulation of cell growth. Perhaps for this reason, alterations at every component of this pathway have been identified in tumor cells and include gene amplification, mutation and deletion, inhibition of transcription by DNA hypermethylation or mRNA stability, and changes in the rate of protein degradation.3 Deletion is the most common mechanism of p16INK4a gene inactivation, having been observed in a wide variety of tumor-derived cell lines and primary tumors.4
REFERENCES
1. Hall, M. et al: Oncogene 11:1581-8, 1995
2. del Arroyo A.G. & Peters, G.: Adv. Exp. Med. Biol. 570:227-47, 2005
3. Kim, W.Y. & Sharpless, N. E.: Cell 127:265-75, 2006
4. Gil, J. & Peters, G.: Nature Rev. Mol. Cell. Biol. 7:667-77, 2006
2. del Arroyo A.G. & Peters, G.: Adv. Exp. Med. Biol. 570:227-47, 2005
3. Kim, W.Y. & Sharpless, N. E.: Cell 127:265-75, 2006
4. Gil, J. & Peters, G.: Nature Rev. Mol. Cell. Biol. 7:667-77, 2006
Products are for research use only. They are not intended for human, animal, or diagnostic applications.
Параметры
Cat.No.: | CG1376 |
Antigen: | Synthesized peptide derived from human p16 INK |
Isotype: | Rabbit IgG |
Species & predicted species cross- reactivity ( ): | Human, Mouse |
Applications & Suggested starting dilutions:* | WB 1:500-1:1000 IP n/d IHC n/d ICC n/d FACS n/d ELISA 1:5000 |
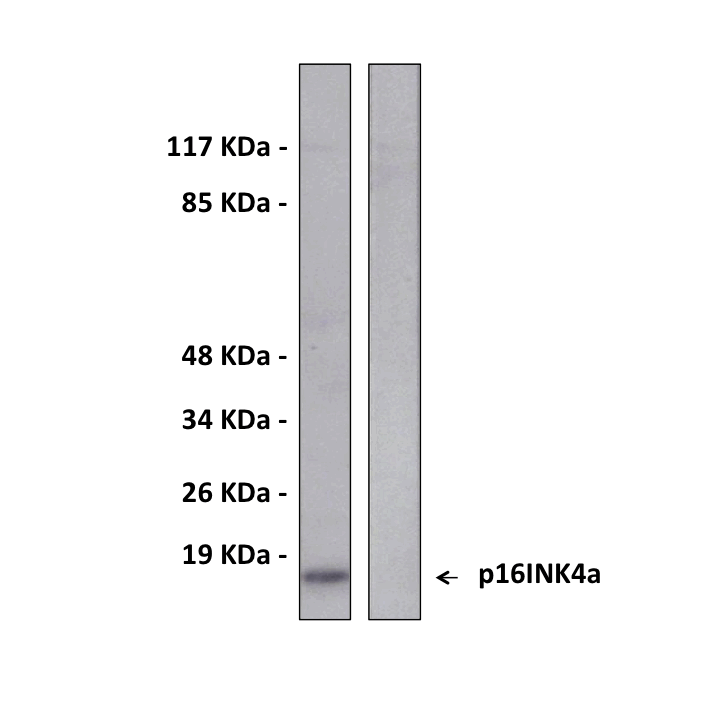
Predicted Molecular Weight of protein: | 16 kDa |
Specificity/Sensitivity: | Detects endogenous p16INK4a proteins without cross-reactivity with other family members. |
Storage: | Store at -20°C, 4°C for frequent use. Avoid repeated freeze-thaw cycles. |
*Optimal working dilutions must be determined by end user.
Документы

Информация представлена исключительно в ознакомительных целях и ни при каких условиях не является публичной офертой



